Critical analysis of antibacterial agents in clinical development
The antibacterial agents currently in clinical development are predominantly derivatives of well-established antibiotic classes and were selected to address the class-specific resistance mechanisms and determinants that were known at the time of their discovery. Many of these agents aim to target the antibiotic-resistant priority pathogens listed by the WHO, including Gram-negative bacteria in the critical priority category, such as carbapenem-resistant Acinetobacter, Pseudomonas and Enterobacterales. Although some current compounds in the pipeline have exhibited increased susceptibility rates in surveillance studies that depend on geography, pre-existing cross-resistance both within and across antibacterial classes limits the activity of many of the new agents against the most extensively drug-resistant (XDR) and pan-drug-resistant (PDR) Gram-negative pathogens. In particular, cross-resistance to unrelated classes may occur by co-selection of resistant strains, thus leading to the rapid emergence and subsequent spread of resistance. There is a continued need for innovation and new-class antibacterial agents in order to provide effective therapeutic options against infections specifically caused by XDR and PDR Gram-negative bacteria.
Similar content being viewed by others
Antibiotics in the clinical pipeline as of December 2022
Article Open access 08 June 2023

The global preclinical antibacterial pipeline
Article 19 November 2019
In vitro and in vivo activity of GT-1, a novel siderophore cephalosporin, and GT-055, a broad-spectrum β-lactamase inhibitor, against biothreat and ESKAPE pathogens
Article Open access 14 September 2021
Introduction
There is a widely acknowledged need for new antibacterial agents to address the global increase in resistance, and this need for new agents is especially urgent for the treatment of antibiotic-resistant Gram-negative bacteria. In early 2017, the WHO convened a group of experts that used a multi-criteria decision analysis method to prioritize the need for new drugs to treat antibiotic-resistant bacteria 1 . The WHO assigned the highest priority to antibacterial drug research and development for the Gram-negative bacteria Acinetobacter, Pseudomonas and species of Enterobacterales that are resistant to carbapenems and are usually extensively drug resistant (XDR) 1 . The same year, the WHO released a clinical pipeline report, which was updated in 2018 and 2019 (refs 2,3 ; WHO clinical pipeline report). The clinical pipeline reports analysed antibiotics and biologics according to their activity against the critical-priority pathogens carbapenem-resistant Acinetobacter baumannii (CRAB), carbapenem-resistant Pseudomonas aeruginosa (CRPA), extended-spectrum β-lactamase (ESBL)-producing Enterobacterales and carbapenem-resistant Enterobacterales (CRE). The level of innovation in the global clinical pipeline was assessed on the basis of the absence of pre-existing cross-resistance to currently used antibacterial drugs 4 .
In this Review, we summarize the current published literature and the publicly available information on antibacterial agents in all phases of clinical development, according to the WHO pipeline report 3 . This Review is limited to antibacterial agents that were in clinical development for systemic human use and that did not yet have regulatory approval anywhere in the world for human use. Additionally, drugs against Clostridioides difficile infection are included, although mostly these agents are not absorbed systemically, because of their oral administration. The principal focus is on the ability of new agents to treat infections caused by bacteria that are XDR or pan-drug-resistant (PDR), the main driver of research and development 5 . Thus, bacteriological information is an important basis for this analysis. We further analyse the gaps in the global clinical pipeline and the need for future antibacterial agents. Although other therapeutic and preventive approaches have been developed, in this Review we focus on direct-acting small molecules for therapeutic purposes (‘traditional antibiotics’). Non-traditional therapies 6,7 and anti-tuberculosis treatments 8,9 have been extensively reviewed elsewhere. Discussing the economic challenges of antibacterial drug development is beyond the scope of this Review.
Top three resistant pathogens
Carbapenem-resistant Acinetobacter baumannii
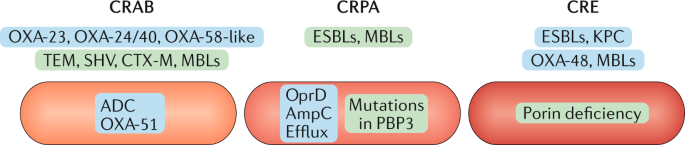
The prevalence of carbapenem resistance among Acinetobacter spp. is extremely variable, ranging from 90% (ref. 10 ). XDR is common because of the diverse and extensive arsenal of chromosomally encoded and acquired resistance genes carried by this pathogen (Fig. 1). Overexpression of the intrinsic chromosomally encoded ß-lactamase ADC (class C β-lactamase in A. baumannii) and of OXA-51-like enzymes, as well as of diverse acquired β-lactamases — such as OXA-23-like, OXA-24/40-like, OXA-58-like and class A β-lactamases (for example, TEM and SHV) — found on a range of mobile genetic elements, is the main resistance mechanism against β-lactam antibiotics 11 . Combinations of different β-lactamases and the accumulation of other resistance determinants, such as aminoglycoside-modifying enzymes, porin deficiencies, efflux or target protein modifications, render CRAB resistant to most of the usually available antibacterial drugs and increasingly also to tigecycline and colistin. XDR and PDR isolates are routinely being reported in some countries 12,13 .
Carbapenem-resistant Pseudomonas aeruginosa
CRPA is genetically diverse and usually exhibits more than one resistance mechanism, including porin deficiency (OprD), hyperproduction of the chromosomally encoded cephalosporinase AmpC, efflux pumps and various class-specific resistance mechanisms (for example, mutations in quinolone resistance-determining regions) 14 . These chromosomally encoded resistance determinants, alone or in combination, affect carbapenems and other β-lactams differently, and diverse combinations are prevalent in isolates from distinct countries 14 . Loss of porins is the most common mechanism leading to carbapenem resistance. Overexpression of one of the several efflux pumps (for example, mexB or mexY) is a nonspecific mechanism that especially affects cefepime, aminoglycosides, fluoroquinolones and meropenem 15 . Acquired resistance mechanisms such as β-lactamases (for example, ESBLs or class B metallo-β-lactamases (MBLs)) are often co-transferred with genes that encode aminoglycoside-modifying enzymes, but they are found less frequently than resistance conferred by chromosomally encoded genes. The prevalence of MBLs, especially VIM and IMP, is highly variable in different countries, and they are becoming increasingly widespread in some regions 14 . The accumulation of several resistance mechanisms to different antibacterial classes can lead to XDR or even PDR strains 16 (Fig. 1).
Carbapenem-resistant Enterobacterales
β-lactam-based clinical pipeline
As is shown by the four recently approved β-lactamase inhibitor (BLI) combinations (two approved since 2017) (Table 1), the clinical pipeline (Table 2) is characterized by new derivatives of the β-lactam class or the functional class of BLIs, most of them focused on improvements in the coverage of Enterobacterales 3 . The renaissance of β-lactam antibiotics in the beginning of the 21st century has enriched the therapy options and addressed class-specific resistance due to the production of β-lactamases by either combining a β-lactam-containing molecule with a BLI or modifying it to prevent hydrolysis.
New stand-alone β-lactam antibiotics
Cefiderocol is a cephalosporin structurally related to ceftazidime and cefepime but more stable to various β-lactamases. The cephalosporin molecule is linked to a siderophore that can bind to iron, which facilitates bacterial cell entry in addition to the usual entry via porin channels; this results in improved penetration of cefiderocol in the bacterial cell, which is most relevant in non-fermenters 61,62 . In Enterobacterales, variations in iron transport channel expression, which varies by species and within species, may cause a wide distribution of MICs 63 . Compared with ceftazidime–avibactam, cefiderocol has similar in vitro microbiological activity against Enterobacterales and P. aeruginosa, but it is more active against Acinetobacter, Stenotrophomonas and Burkholderia species. The resistance prevalence in CRE and CRPA is a little lower than with ceftazidime–avibactam 64 . The main weakness of cefiderocol is its limited activity against NDM-producing E. coli as well as OXA-23-producing and OXA-24-producing Acinetobacter spp 65 . MIC90 values are about eight doubling dilutions higher in KPC-2-producing Enterobacterales than in wild-type strains. Due to the very low MICs in wild-type strains, the reduction of activity may still keep cefiderocol MICs for many strains below the clinical breakpoint 63 . Similarly, the reduced susceptibility of ESBL-producing, carbapenemase-producing or AmpC-producing strains may not translate into immediate clinical resistance 63 but may be problematic in critically ill patients with altered pharmacokinetic profiles. However, the most problematic XDR or PDR strains have variable susceptibility, with ~64% of NDM-producing pathogens testing as susceptible 65 . MICs clustering around the breakpoint may be the beginning of a stepwise development of resistance, and reliable susceptibility testing will be essential.
Cefiderocol has recently been approved by the FDA for the treatment of complicated UTIs (including pyelonephritis) caused by susceptible Gram-negative bacteria in patients with limited or no alternative treatment options 66 . However, the results from a small descriptive study in critically ill patients with XDR Gram-negative bacteria have raised concerns, as higher rates of infection-related and non-infection-related deaths with treatment failure were seen for cefiderocol than for the comparator (best available therapy). In addition, the development of cefiderocol resistance during therapy was linked to poor outcomes. The results of this study, with its inherent limitations of a small descriptive trial, leave clinicians with little evidence to support the use of cefiderocol in patients with XDR Gram-negative bacteria, especially Acinetobacter spp. and Pseudomonas spp. 67 . A better understanding of cefiderocol efficacy and the associated risk factors will require additional well-designed studies.
BOS-228 is a new monobactam in phase II clinical development. Like other monobactams, BOS-228 inhibits PBP3 and is stable to MBLs. Additionally, it has been modified to resist hydrolysis by most serine β-lactamases in Enterobacteriaceae 68 . Cross-resistance exists with aztreonam when non-β-lactamase mechanisms are involved (for example, upregulated efflux and/or porin downregulation, or mutations in PBP3 and the envelope stress response system) 69 .
Sulopenem and tebipenem, in the form of bioavailable esters, are both in phase III clinical development and will provide new oral therapies for ESBL-producing (MDR) Enterobacteriaceae, but they also confront the medical community with specific challenges. Sulopenem is a thiopenem with intravenous and oral formulations (prodrug), whereas tebipenem is an orally available carbapenem (as a prodrug) that was approved in Japan in 2009 for the treatment of paediatric respiratory tract infections (therefore, it is not included in Table 2). Both antibiotics have similar spectra against Gram-negative bacteria compared with ertapenem, which focuses on ESBL-producing Enterobacteriaceae. These agents show complete cross-resistance with ertapenem and other carbapenems 70 . Sulopenem failed to demonstrate non-inferiority compared with ertapenem for complicated intra-abdominal infections in a recent phase III trial 71 . Sulopenem and tebipenem were developed for community-acquired uncomplicated UTIs and for oral follow-on after intravenous therapy for complicated UTIs 72,73 . If used widely, they will most likely exert a strong selection pressure for carbapenem resistance, like any other carbapenem 74 . Antibiotic stewardship programmes would need to be established in the community to mitigate this risk. From the public health perspective, oral carbapenem-sparing options for the treatment of MDR strains would be preferable.
Tetracycline derivatives
New semisynthetic and synthetic tetracycline derivatives, based on new chemistry approaches, have been recently approved or are in clinical development. Tetracyclines were discovered in the 1940s and have been widely used in human and veterinary medicine and in agricultural applications, which has contributed to the widespread dissemination of tetracycline resistance. Efflux pumps and ribosome protection proteins are the most common resistance mechanisms 75 . In Gram-negative bacteria, target-site mutations have been described, recently even for tigecycline 76 . Lately, enzymatic degradation has gained more attention, as new, highly transferrable plasmid-mediated tetracycline-inactivating enzymes, Tet(X3) and Tet(X4), have been described and are globally present. They degrade all tetracyclines, including tigecycline and the recently approved eravacycline and omadacycline 77,78,79 . Such plasmids may carry other resistance determinants, such as mcr1 (colistin resistance) and genes encoding MBLs (for example, NDM), thus potentially rendering isolates XDR 78,80 . The fully synthetic eravacycline (intravenous) has activity against CRE comparable to tigecycline and exhibits cross-resistance with tigecycline 79,81 . The semisynthetic minocycline derivative omadacycline has been recently approved for the oral and intravenous treatment of acute bacterial skin and skin structure infections (ABSSSI) and community-acquired pneumonia (CAP). The activity against XDR Enterobacterales is insufficient 82 .
Three more tetracyclines are in early clinical development: first is KBP-7022, with a spectrum similar to tigecycline 83,84 but lower MICs in Acinetobacter spp. 85 . No information is available about cross-resistance with tigecycline. The second tetracycline is TP-6076, with activity against Enterobacterales and Acinetobacter spp. and lower MICs than tigecycline. The impact of these in vitro findings is not known yet, pending pharmacokinetic–pharmacodynamic studies and breakpoint decisions. In tigecycline-non-susceptible and minocycline-non-susceptible Acinetobacter isolates, the MICs of TP-6076 were slightly increased 86 . In K. pneumoniae, tigecycline-resistant strains are mostly also resistant to TP-6076 (ref. 87 ), and resistance to tigecycline in carbapenem-resistant strains is high in some areas 27 . Resistance in K. pneumoniae is due to overexpressed RamA, a transcriptional regulator that modulates efflux pump expression 88 . Such a resistance mechanism is increasingly found in CRE with porin deficiency, production of various β-lactamases and aminoglycoside-inactivating enzymes 28 , thus reducing the value of this antibiotic as a treatment option for the most resistant bacteria. It is not yet known whether the liabilities of tigecycline, including low plasma exposure, concentration-independent plasma protein binding and adverse-event concerns, will be mirrored by the new tetracyclines 89 . Preliminary information points to an adverse-event profile of TP-6067 similar to that of tigecycline, with dose-dependent gastrointestinal side effects, including nausea and vomiting as the most frequently reported adverse effects. The third tetracycline — TP-271, a synthetic tetracycline — is very similar to tigecycline and is active against respiratory pathogens. It is not affected by Tet(M) (ribosomal protection protein), but is affected by Tet(A) (efflux) and Tet(X) (enzymatic inactivation) 90 .
Similar to other derivatives of long-used classes, the new tetracyclines address some class-specific resistance mechanisms of the older representatives of this class, such as doxycycline, but have a high degree of cross-resistance to tigecycline. They do not represent reliable alternatives for XDR or PDR Gram-negative bacteria. Potential benefits over tigecycline regarding non-potency-related characteristics, such as pharmacokinetics and safety, have not been shown so far.
Other derivatives of old classes
In addition to β-lactams, β-lactamase inhibitors and tetracyclines, a few other old classes have been used as starting points to develop derivatives. The polymyxins are seeing a revival with SPR206, a polymyxin derivative with slightly improved potency that also reduces the magnitude of cross-resistance. The increasingly described plasmid-mediated mcr resistance is often associated with other resistance genes that encode for β-lactamases (including carbapenemase) and for resistances to other antibacterial classes, which may include fluoroquinolones and aminoglycosides 91 . Thus, several antibiotic classes may select for XDR or even PDR strains. Carbapenem-resistant K. pneumoniae, which also exhibited resistance to colistin in more than 30% of the strains, has been described in a recent study 28 . It is not clear yet how polymyxin resistance and associated cross-resistance to unrelated classes will develop over the next years, whether SPR206 will be of any benefit in polymyxin-resistant strains and whether the promise of lower class-specific toxicity will hold true in patients 92 .
Another polymyxin derivative (SPR741), with limited intrinsic activity but improved safety, is being studied for permeabilization of the outer membrane, thereby granting antibacterial agents access to their intracellular targets 93 . The most beneficial combinations of such a ‘potentiator’ strategy are not known yet, and development is pending.
The aminoglycoside class has seen a recent addition, plazomicin, to reduce the impact of aminoglycoside-modifying enzymes. Although plazomicin has activity against many CRE isolates, organisms that produce the NDM-1 MBL or OXA-48 enzymes are often resistant to plazomicin, due to co-production of ribosomal methyl transferase 94,95 . Another aminoglycoside has also entered its first trials in humans. This aminoglycoside, apramycin, has been licensed since 1980 for oral use in veterinary medicine. It was active in 87% of carbapenem-resistant Klebsiella isolates from Greek hospitals, a susceptibility rate similar to that for plazomicin. Though apramycin is not affected by 16S rRNA methylases that cause resistance to plazomicin and all other aminoglycosides, it is ineffective against the acetyltransferase AAC(3)-IV-producing strains 96,97 . Resistance in human isolates was already described in 1993 (ref. 98 ). Its usefulness against XDR Gram-negative strains will depend on the future epidemiology of aminoglycoside resistance, but rapid resistance development and spread could be anticipated if apramycin were to be used clinically.
Derivatives of old classes with activity against Gram-positive bacteria are in clinical development. No information on their differentiation from the drugs already available has been published concerning the oxazolidinone contezolid, with equal activity and non-inferiority compared with linezolid 99 , and the ketolide nafithromycin, which is comparable to telithromycin 100 . Two rifamycin conjugates, TNP-2092 (rifamycin–quinolizinone conjugate with rifampicin-like activity 101 ) and TNP-2198 (rifamycin–nitroimidazole conjugate) are in clinical development. The published information has been insufficient to assess the potential clinical usefulness of these rifamycin conjugates.
Beyond old classes
New chemical scaffolds, new targets or binding sites, and a new mode of action
Two new antibiotics that represent new chemical scaffolds with activity against Gram-positive bacteria are in clinical development. The target FabI, an enoyl-ACP reductase that is the rate-limiting enzyme in the last step for fatty-acid biosynthesis, is known from existing FabI inhibitors (isoniazid, Mycobacterium tuberculosis) and triclosan (in some consumer products) 102 . Afabicin (Debio-1450) is a new intravenously and orally administered FabI inhibitor that exhibits selective antibacterial activity against staphylococcal species 103 . As expected, there is no cross-resistance with other antibacterial drugs used for staphylococcal infections 104 . Preliminary studies have indicated that afabicin may not be prone to rapid emergence of resistance, despite binding to a single target, possibly due to its high-affinity binding 102 . It is currently being developed for the treatment of bone and joint infections 105 . Other FabI inhibitors are in preclinical development (for example, MUT056399) 106 .
The prodrug TXA709 is a benzamide compound and targets the bacterial protein FtsZ, which has an essential role in septum formation and prevents bacterial cell division without a eukaryotic homologue 107 . Its activity is focused on S. aureus, with no pre-existing cross-resistance to commonly used antibiotics, due to its new chemical class, new target and new mode of action. As expected, this single-target agent leads to a relatively high frequency of resistance, which may be mitigated by using it in combination with other agents 108 . Though the immediate medical need for a new drug against multiresistant S. aureus is low, a new-class antibacterial drug may be valuable in selected cases and on a broader basis in the future.
The topoisomerase inhibitors zoliflodacin (spiropyrimidinetrione) and gepotidacin (triazaacenaphthylene) are new chemical scaffolds and bind to gyrase, the same target as for the fluoroquinolones, but at distinct binding sites. They were developed for the treatment of uncomplicated urogenital gonorrhoea. Due to its different chemical structure and distinct binding site, zoliflodacin has so far not shown to be cross-resistant to fluoroquinolones 109 . For gepotidacin, a phase II study in uncomplicated urogenital gonorrhoea raised some questions regarding cross-resistance to ciprofloxacin and the emergence of resistance. Three isolates with higher gepotidacin MICs were quinolone resistant and showed a mutation in the parC gene, which is known to affect gepotidacin binding. Two of these isolates developed high-level resistance to gepotidacin and were bacteriological failures in the clinical trial 110 . Due to overlapping binding sites, gepotidacin may show some cross-resistance to ciprofloxacin, and resistance may soon emerge once it is used in clinical practice. Gepotidacin is currently also being developed for uncomplicated UTIs. Its MICs against fluoroquinolone-susceptible E. coli are much higher than for levofloxacin, but cross-resistance to fluoroquinolones has not been described yet in E. coli 111,112 .
Although fluoroquinolones target both the DNA gyrase GyrA subunit and the topoisomerase IV ParC subunit, no inhibitor of GyrB and/or ParE is currently in clinical use 113 . The new GyrB inhibitor SPR-720 (aminobenzimidazole) inhibits the ATPase activity of gyrase in M. tuberculosis and non-tuberculous mycobacteria (NTM) 114 . It will be developed for these bacteria.
The development of intravenous murepavidin, a cyclic peptide that targets the lipopolysaccharide transport protein D (LptD) in P. aeruginosa, was terminated in July 2019 owing to concerns about nephrotoxicity observed in phase III. The inhaled form of murepavidin is in preclinical development for P. aeruginosa infections in patients with cystic fibrosis. Therefore, it is not included in Table 2.
Lefamulin, a member of the pleuromutilin class, was approved by the FDA for the treatment of community-acquired bacterial pneumonia in 2019 (ref. 115 ). Though pleuromutilins are an established class for systemic use in veterinary medicine and have been used topically in humans (retapamulin was approved in 2007), lefamulin represents a new scaffold for systemic use in humans. Whether the prior use and selection pressure of this class will accelerate the emergence of resistance against lefamulin remains to be seen. Caution should be exercised when prescribing this drug to patients with QT prolongation and ventricular arrhythmias. It has shown embryo-fetal toxicity in animals and should not be prescribed to pregnant women and females of reproductive potential without effective contraception 115 .
New antibacterial agents against Clostridioides difficile
New antibacterial drugs against C. difficile infection are being developed with the goal of reducing recurrences. These agents are usually orally available and not absorbed, and thus systemic pharmacokinetics and toxicity do not represent major challenges in the discovery process. This group of antibiotics has been included because they represent mainly new chemical classes with new targets and new modes of action. The most advanced compound is ridinilazole, a bis-benzimidazole that has been suggested to inhibit cell division and is associated with reduction in spore and toxin production 116,117 . Three new chemical structures with new targets and new modes of action are in early clinical development. MGB-BP3 is a distamycin derivative and binds to the DNA minor groove 118 . It acts on multiple binding sites and interferes with transcription. ACX-362E is a synthetic purine and targets DNA polymerase IIIC 119 . CRS-3123 is a diaryldiamine derivative that inhibits the Met-aminoacyl-tRNA synthetase 120 . Little information is available about the propensity for rapid emergence of single-step resistance due to mutations in the target of CRS-3123. The clinical value of these new drugs will depend on proof that they can reliably reduce the rate of recurrence.
Conclusion
This narrative overview has critically reviewed the antibacterial agents in clinical development and confirms the limited scope of these new antibacterial agents, especially against Gram-negative critical-priority pathogens. In particular, all agents in development against the critical-priority pathogens exhibit some degree of pre-existing cross-resistance. A summary of our conclusions may be found in Box 1.
The global clinical pipeline is dominated by derivatives of known chemical and functional classes. Though the experience with widely used antibacterial classes and familiarity with class-specific safety profiles and pharmacokinetic and pharmacodynamic properties are good starting points for antibacterial research and development programmes, chemical modifications lead to improvements that are usually incremental and address only selected class-specific resistance mechanisms that are known at the time of lead optimization, whereas other mechanisms remain unaffected. Thus, a relatively high rate of pre-existing cross-resistance in XDR strains or substantially increased MICs compared with the wild-type strains may limit the benefit of such new therapies in many geographic regions and locations. They are unlikely to ease the global threat of XDR and PDR Gram-negative bacteria. Knowledge of the surveillance data on the distribution of molecular epidemiology and resistance mechanisms at the local level will be an absolute prerequisite for adequate therapy decisions. This fact, and the focus on one or a few specific pathogens, limits these antibacterial drugs for use in the initial treatment phase of critically ill patients, before pathogen and susceptibility profiles are known.
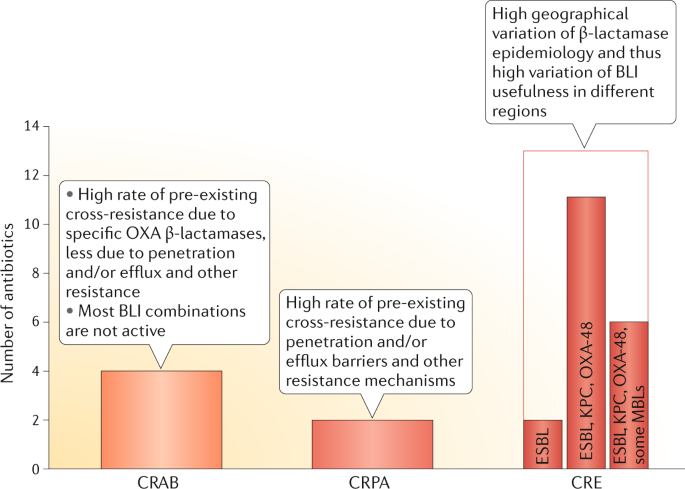
Most new drugs in clinical development are not active against CRAB. The three compounds that are active (a BLI combination, a polymyxin derivative and a tetracycline derivative) are all affected by pre-existing cross-resistance or by substantially increased MICs compared with wild-type strains, which may reduce their efficacy in critically ill patients due to pharmacokinetic and pharmacokinetic–pharmacodynamic variability 121 . In Pseudomonas spp., nonspecific resistance mechanisms such as porin deficiency and overexpressed efflux pumps may lead to a high baseline resistance rate to new derivatives and reduce the usefulness not only of new BLI combinations but also of other antibacterial classes.
In addition to pre-existing cross-resistance in all key bacterial pathogens, new resistance will emerge rapidly to newly introduced derivatives if they are used widely and replace older empiric therapies without adequate stewardship. The emergence of new resistance determinants is also driven by all currently used antibiotics of the old class. Some of these examples have already been described for ceftazidime–avibactam, other BLI combinations, new tetracyclines and plazomicin. Unpredictable components of the evolution of resistance to known antibacterial classes 122 may increase the risk of unacceptably high rates of resistance to a new derivative that emerge during development or soon thereafter. Though discovering and developing new chemical structures for new bacterial targets is extremely challenging, it is the most promising strategy for starting with an effective antibacterial agent without pre-existing cross-resistance. Substantial efforts should be directed to this endeavour.
The antibacterial drugs currently in phase III clinical development that target Gram-negative bacteria are usually evaluated for the treatment of complicated UTIs, complicated intra-abdominal infections (cIAI), and sometimes hospital-acquired pneumonia (HAP) and ventilator-associated pneumonia (VAP) caused by susceptible bacteria. They are compared to standard-of-care antibiotics in non-inferiority clinical trial designs. Though complicated UTI and cIAI caused by susceptible bacteria are not a high unmet medical need, as several antibacterial alternatives exist, this development strategy is aligned with a streamlined regulatory pathway that ensures a more robust safety population. When a new antibacterial drug is approved, clinicians often have minimal information about its efficacy in patients infected with XDR or PDR bacteria, where the medical need is highest, even when clinical trials are designed to include such patients. In addition to the streamlined pivotal studies undertaken to achieve approval, only company-sponsored non-clinical information is available to provide some early indications of potential differentiations between similar drugs. After approval, physicians are faced with a lack of independent critical analysis of the data and evidence, a lack of surveillance systems to rapidly detect new patterns of resistance, a lack of updated therapeutic guidelines and a lack of rapid diagnostic capabilities in many institutions. Nevertheless, these new antibiotics may still be useful for individual patients or specific situations, even without the availability of needed evidence at the time of approval based on careful situation-specific evaluation of the new drug.
All the new-class antibacterial agents focus on Gram-positive bacteria (especially S. aureus or C. difficile) or Gram-negative cocci (for example, Neisseria gonorrhoeae), thus highlighting the scientific barriers to antibiotic discovery in the field of Gram-negative rods. Overcoming barriers to drug penetration and efflux avoidance for Gram-negative bacteria is still a main hurdle that impedes innovation 123 .
In conclusion, the need for research and development of new antibacterial drugs, especially against the WHO critical-priority pathogens, is still strong. Efforts should focus on innovation and antibacterial agents without pre-existing cross-resistance (that is, on new classes or new targets) in order to provide drugs for the most resistant pathogens and prepare for unpredictable resistance challenges in the future. The discovery of such new antibiotics will require sustained commitment over a long time period; substantial levels of resources will be needed to solve the numerous challenges of new-class antibacterial agents, and collective efforts will be needed to expand the science base.
Box 1 Antibacterial drugs in clinical development and future perspectives
- The clinical pipeline is dominated by derivatives of most major known chemical and functional classes, especially β-lactams and β-lactamase inhibitors (BLIs), but also tetracyclines.
- All antibiotic candidates currently under development to treat infections caused by the WHO critical-priority bacteria have at least some cross-resistance with existing agents.
- Selected class-specific resistance mechanisms are being addressed, others remain unaffected.
- The rate of cross-resistance is relatively high in extensively drug-resistant (XDR) strains and specifically in pan-drug-resistant (PDR) strains.
- The pipeline focused on carbapenem-resistant Enterobacterales (CRE) has incomplete β-lactamase coverage, especially for β-lactamases prevalent in Asia and Africa, but also in some European countries.
- Susceptibility rates depend on the epidemiology of resistance mechanisms in different regions and locations.
- The best use of new drugs is achieved if the regional molecular resistance epidemiology is known and if agents are selected according to susceptibility tests.
- Few innovative new antibiotics are being developed against Staphylococcus aureus, as well as Neisseria gonorrhoeae and Clostridioides difficile.
- There is a continued high need for innovation, especially for new-class antibiotics without pre-existing cross-resistance.
References
- Tacconelli, E. et al. Discovery, research, and development of new antibiotics: the WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis.18, 318–327 (2018). This WHO list prioritizes target pathogens for antibacterial drug research and discovery with a global perspective. PubMedGoogle Scholar
- Theuretzbacher, U. et al. Analysis of the clinical antibacterial and antituberculosis pipeline. Lancet Infect. Dis.19, e40–e50 (2019). CASPubMedGoogle Scholar
- World Health Organization. 2019 antibacterial agents in clinical development: an analysis of the antibacterial clinical development pipeline. https://apps.who.int/iris/bitstream/handle/10665/330420/9789240000193-eng.pdf (WHO, 2019).This WHO report presents the updated analysis of the global clinical pipeline including drugs against tuberculosis.
- Theuretzbacher, U. Antibiotic innovation for future public health needs. Clin. Microbiol. Infect.23, 713–717 (2017). CASPubMedGoogle Scholar
- Theuretzbacher, U. Global antimicrobial resistance in Gram-negative pathogens and clinical need. Curr. Opin. Microbiol.39, 106–112 (2017). CASPubMedGoogle Scholar
- Theuretzbacher, U. & Piddock, L. J. V. Non-traditional antibacterial therapeutic options and challenges. Cell Host Microbe26, 61–72 (2019). CASPubMedGoogle Scholar
- Rex, J. H., Fernandez Lynch, H., Cohen, I. G., Darrow, J. J. & Outterson, K. Designing development programs for non-traditional antibacterial agents. Nat. Commun.10, 3416–3416 (2019). PubMedPubMed CentralGoogle Scholar
- J Libardo, M. D., Boshoff, H. I. & Barry, C. E. III. The present state of the tuberculosis drug development pipeline. Curr. Opin. Pharmacol.42, 81–94 (2018). CASPubMedGoogle Scholar
- World Health Organization. Global tuberculosis report 2019. https://www.who.int/tb/publications/global_report/en/ (WHO, 2019).
- Xie, R., Zhang, X. D., Zhao, Q., Peng, B. & Zheng, J. Analysis of global prevalence of antibiotic resistance in Acinetobacter baumannii infections disclosed a faster increase in OECD countries. Emerg. Microbes Infect.7, 31 (2018). PubMedPubMed CentralGoogle Scholar
- Wright, M. S. et al. New insights into dissemination and variation of the health care-associated pathogen Acinetobacter baumannii from genomic analysis. mBio5, e00963-13 (2014). Acinetobactershows widespread genetic variation among strains, a high variability in antibiotic resistance determinants and dynamic genomic change over short evolutionary time spans. PubMedPubMed CentralGoogle Scholar
- Asif, M., Alvi, I. A. & Rehman, S. U. Insight into Acinetobacter baumannii: pathogenesis, global resistance, mechanisms of resistance, treatment options, and alternative modalities. Infect. Drug. Resist.11, 1249–1260 (2018). CASPubMedPubMed CentralGoogle Scholar
- Tuan Anh, N. et al. Molecular epidemiology and antimicrobial resistance phenotypes of Acinetobacter baumannii isolated from patients in three hospitals in southern Vietnam. J. Med. Microbiol.66, 46–53 (2017). PubMedGoogle Scholar
- Castanheira, M., Deshpande, L. M., Costello, A., Davies, T. A. & Jones, R. N. Epidemiology and carbapenem resistance mechanisms of carbapenem-non-susceptible Pseudomonas aeruginosa collected during 2009–11 in 14 European and Mediterranean countries. J. Antimicrob. Chemother.69, 1804–1814 (2014). CASPubMedGoogle Scholar
- Cabot, G. et al. Overexpression of AmpC and efflux pumps in Pseudomonas aeruginosa isolates from bloodstream infections: prevalence and impact on resistance in a Spanish multicenter study. Antimicrob. Agents Chemother.55, 1906–1911 (2011). CASPubMedPubMed CentralGoogle Scholar
- del Barrio-Tofiño, E. et al. Genomics and susceptibility profiles of extensively drug-resistant Pseudomonas aeruginosa isolates from Spain. Antimicrob. Agents Chemother.61, e01589–17 (2017). This resistome analysis of extensively drug-resistantPseudomonasreveals the molecular epidemiology and a broad variety of resistance mechanisms. PubMedPubMed CentralGoogle Scholar
- Sader, H. S., Flamm, R. K., Streit, J. M., Doyle, T. B. & Castanheira, M. Antimicrobial activity of cefepime–zidebactam (WCK 5222) against clinical isolates of carbapenem-resistant Enterobacterales collected worldwide in 2018. ASM/ESCMID Drug Development Conference 2019https://www.jmilabs.com/data/posters/ASM-ESCMID2019-cefepime-zidebactam-CRE.pdf (2019).
- Castanheira, M., Doyle, T. B., Kantro, V., Mendes, R. E. & Shortridge, D. Meropenem–vaborbactam activity against carbapenem-resistant Enterobacterales isolates collected in U.S. hospitals during 2016–2018. Antimicrob. Agents Chemother.64, 01951–19 (2019). Google Scholar
- Dadashi, M. et al. Frequency distribution, genotypes and the most prevalent sequence types of New Delhi metallo-beta-lactamase-producing Escherichia coli among clinical isolates around the world; a review. J. Glob. Antimicrob. Resist.19, 284–293 (2019). PubMedGoogle Scholar
- Bush, K. & Bradford, P. A. Epidemiology of beta-lactamase-producing pathogens. Clin. Microbiol. Rev. 33, e00047-19 (2020). PubMedGoogle Scholar
- Grundmann, H. et al. Occurrence of carbapenemase-producing Klebsiella pneumoniae and Escherichia coli in the European survey of carbapenemase-producing Enterobacteriaceae (EuSCAPE): a prospective, multinational study. Lancet Infect. Dis.17, 153–163 (2017). CASPubMedGoogle Scholar
- Mueller, L. et al. Phenotypic, biochemical and genetic analysis of KPC-41, a KPC-3 variant conferring resistance to ceftazidime–avibactam and exhibiting reduced carbapenemase activity. Antimicrob. Agents Chemother.63, 01111–01119 (2019). Google Scholar
- Giddins, M. J. et al. Successive emergence of ceftazidime–avibactam resistance through distinct genomic adaptations in bla(KPC-2)-harboring Klebsiella pneumoniae sequence type 307 isolates. Antimicrob. Agents Chemother.62, e02101–e02117 (2018). PubMedPubMed CentralGoogle Scholar
- Palzkill, T. Structural and mechanistic basis for extended-spectrum drug-resistance mutations in altering the specificity of TEM, CTX-M, and KPC β-lactamases. Front. Mol. Biosci.5, 16–16 (2018). PubMedPubMed CentralGoogle Scholar
- Chetri, S. et al. AcrAB-TolC efflux pump system plays a role in carbapenem non-susceptibility in Escherichia coli. BMC Microbiol.19, 210 (2019). PubMedPubMed CentralGoogle Scholar
- Alm, R. A., Johnstone, M. R. & Lahiri, S. D. Characterization of Escherichia coli NDM isolates with decreased susceptibility to aztreonam/avibactam: role of a novel insertion in PBP3. J. Antimicrob. Chemother.70, 1420–1428 (2015). CASPubMedGoogle Scholar
- Galani, I. et al. Nationwide epidemiology of carbapenem resistant Klebsiella pneumoniae isolates from Greek hospitals, with regards to plazomicin and aminoglycoside resistance. BMC Infect. Dis.19, 167 (2019). PubMedPubMed CentralGoogle Scholar
- Capone, A. et al. High rate of colistin resistance among patients with carbapenem-resistant Klebsiella pneumoniae infection accounts for an excess of mortality. Clin. Microbiol. Infect.19, E23–E30 (2013). CASPubMedGoogle Scholar
- Andrey, D. O. et al. An emerging clone, Klebsiella pneumoniae carbapenemase 2-producing K. pneumoniae Sequence Type 16, associated with high mortality rates in a CC258-endemic setting. Clin. Infect. Dis. https://doi.org/10.1093/cid/ciz1095 (2019). ArticlePubMedGoogle Scholar
- David, S. et al. Epidemic of carbapenem-resistant Klebsiella pneumoniae in Europe is driven by nosocomial spread. Nat. Microbiol.4, 1919–1929 (2019). CASPubMedGoogle Scholar
- Pérez-Vázquez, M. et al. Emergence of NDM-producing Klebsiella pneumoniae and Escherichia coli in Spain: phylogeny, resistome, virulence and plasmids encoding blaNDM-like genes as determined by WGS. J. Antimicrob. Chemother.74, 3489–3496 (2019). PubMedGoogle Scholar
- Krajnc, A., Lang, P. A., Panduwawala, T. D., Brem, J. & Schofield, C. J. Will morphing boron-based inhibitors beat the β-lactamases? Curr. Opin. Chem. Biol.50, 101–110 (2019). CASPubMedPubMed CentralGoogle Scholar
- Tehrani, K. H. M. E. & Martin, N. I. β-lactam/β-lactamase inhibitor combinations: an update. MedChemComm9, 1439–1456 (2018). CASPubMedPubMed CentralGoogle Scholar
- Linciano, P., Cendron, L., Gianquinto, E., Spyrakis, F. & Tondi, D. Ten years with New Delhi metallo-beta-lactamase-1 (NDM-1): from structural insights to inhibitor design. ACS Infect. Dis.5, 9–34 (2019). CASPubMedGoogle Scholar
- Lomovskaya, O. et al. Vaborbactam: spectrum of beta-lactamase inhibition and impact of resistance mechanisms on activity in Enterobacteriaceae. Antimicrob. Agents Chemother.61, e01443-17 (2017). PubMedPubMed CentralGoogle Scholar
- Papp-Wallace, K. M. et al. Relebactam is a potent inhibitor of the KPC-2 β-lactamase and restores imipenem susceptibility in KPC-producing Enterobacteriaceae. Antimicrob. Agents Chemother.62, e00174-18 (2018). PubMedPubMed CentralGoogle Scholar
- Karlowsky, J. A. et al. In vitro activity of imipenem/relebactam against Gram-negative ESKAPE pathogens isolated in 17 European countries: 2015 SMART surveillance programme. J. Antimicrob. Chemother.73, 1872–1879 (2018). PubMedGoogle Scholar
- Balabanian, G., Rose, M., Manning, N., Landman, D. & Quale, J. Effect of porins and blaKPC expression on activity of imipenem with relebactam in Klebsiella pneumoniae: can antibiotic combinations overcome resistance? Microb. Drug. Resist.24, 877–881 (2018). CASPubMedGoogle Scholar
- Moussa, S. H., Shapiro, A. B., McLeod, S. M. & A. Miller, A. A. Resistance to sulbactam–durlobactam in clinical isolates of Acinetobacter baumannii is rare and maps to PBP3. Acinetobacter Conference 2019https://www.entasistx.com/application/files/3115/6959/0903/Moussa_SUL_Resistance_Acinetobacter2019_FINAL.pdf. (2019).
- McLeod, S. M. et al. In vitro antibacterial activity of sulbactam–durlobactam (ETX2514) against 121 recent Acinetobacter baumannii isolates from patients in India. IDweek 2019https://www.entasistx.com/application/files/2215/7115/9465/IDweek_2019_ETX2514_India_V11.pdf (2019).
- Barnes, M. D. et al. targeting multidrug-resistant Acinetobacter spp.: sulbactam and the diazabicyclooctenone β-lactamase inhibitor ETX2514 as a novel therapeutic agent. mBio10, e00159-19 (2019). PubMedPubMed CentralGoogle Scholar
- Livermore, D. M., Mushtaq, S., Warner, M. & Woodford, N. Activity of OP0595/β-lactam combinations against Gram-negative bacteria with extended-spectrum, AmpC and carbapenem-hydrolysing β-lactamases. J. Antimicrob. Chemother.70, 3032–3041 (2015). CASPubMedGoogle Scholar
- Mushtaq, S., Vickers, A., Woodford, N., Haldimann, A. & Livermore, D. M. Activity of nacubactam (RG6080/OP0595) combinations against MBL-producing Enterobacteriaceae. J. Antimicrob. Chemother.74, 953–960 (2018). Google Scholar
- Okujava, R. et al. Activity of meropenem/nacubactam combination against Gram-negative clinical isolates: ROSCO global surveillance 2017. Open Forum Infect. Dis.5, S416–S416 (2018). PubMed CentralGoogle Scholar
- Khan, Z., Iregui, A., Landman, D. & Quale, J. Activity of cefepime/zidebactam (WCK 5222) against Enterobacteriaceae, Pseudomonas aeruginosa and Acinetobacter baumannii endemic to New York City medical centres. J. Antimicrob. Chemother.74, 2938–2942 (2019). PubMedGoogle Scholar
- Thomson, K. S., AbdelGhani, S., Snyder, J. W. & Thomson, G. K. Activity of cefepime-zidebactam against multidrug-resistant (MDR) Gram-negative pathogens. Antibiotics8, 32 (2019). CASPubMed CentralGoogle Scholar
- Krajnc, A. et al. Bicyclic boronate VNRX-5133 inhibits metallo- and serine-β-lactamases. J. Med. Chem.62, 8544–8556 (2019). CASPubMedPubMed CentralGoogle Scholar
- Liu, B. et al. Discovery of taniborbactam (VNRX-5133): a broad-spectrum serine- and metallo-β-lactamase inhibitor for carbapenem-resistant bacterial infections. J. Med. Chem. https://doi.org/10.1021/acs.jmedchem.9b01518 (2019).
- Hackel, M. & Sahm, D. Antimicrobial activity of cefepime in combination with VNRX-5133 against a collection of β-lactamase-producing Enterobacteriaceae. ECCMID 2019https://www.ihma.com/app/uploads/VenatoRx_P50_FEP-5133-BL_ECCMID-2019_FINAL.pdf (2019).
- Hackel, M. & Sahm D. Antimicrobial activity of cefepime in combination with VNRX-5133 against a global 2018 surveillance collection of Pseudomonas aeruginosa. ASM Microbe 2019https://www.venatorx.com/wp-content/uploads/2019/07/ASMMicrobe2019-SUNDAY-AAR-722.pdf (2019).
- Castanheira, M., Lindley, J., Huynh, H., Mendes, R. E. & Lomovskaya, O. Activity of novel β-lactamase inhibitor QPX7728 combined with β-lactam agents when tested against carbapenem-resistant Enterobacteriaceae (CRE) isolates. Open. Forum Infect. Dis.6, S309–S309 (2019). PubMed CentralGoogle Scholar
- Mendes,R. E., Rhomberg, P. R., Watters, A. A., Castanheira, M., Flamm, R. K. In vitro activity of the orally bioavailable ceftibuten/VNRX-7145 combination against a challenge set of Enterobacteriaceae pathogens carrying molecularly characterized β-lactamase genes. ECCMID 19https://www.jmilabs.com/data/posters/ECCMID19-VNRX-7145-ceftibuten.pdf (2019).
- John, K. J., Chatwin, C. L., Hamrick, J. C., Moeck, G. & Pevear, D. C. Rescue of ceftibuten activity by the oral β-lactamase inhibitor VNRX-7145 against Enterobacteriaceae expressing class A, C and/or D β-lactamases. ASM Microbe 2019https://www.venatorx.com/wp-content/uploads/2019/07/ASMMicrobe2019-SUNDAY-AAR-719.pdf (2019).
- M. Hackel & Sahm, D. In vitro activity of ceftibuten in combination with VNRX-7145 and comparators against 1,066 UTI isolates non-susceptible to amoxicillin–clavulanate and levofloxacin microbe. ASM Microbe 2019https://www.venatorx.com/wp-content/uploads/2019/07/ASMMicrobe2019-SUNDAY-AAR-721.pdf (2019).
- McLeod, S. et al. The antibacterial activity of sulbactam and the novel b-lactamase Inhibitor ETX2514 combined with imipenem or meropenem against recent clinical isolates of Acinetobacter baumannii and Pseudomonas aeruginosa. ASM Microbe 2017https://www.entasistx.com/application/files/2815/1846/7310/McLeod-et-al-ASM-Microbe-2017-FRI-82.pdf (2017).
- Duncan, L. R., Rhomberg, P. R., Mendes, R. E., Flamm, R. K. & Trias, J. Ceftibuten-avibactam activity against β-lactam-resistant Enterobacteriaceae clinical isolates. ASM Microbe 2019https://www.jmilabs.com/data/posters/ASM-Microbe19-ceftibuten-avibactam.pdf (2019).
- Papp-Wallace, K. M. et al. Beyond piperacillin-tazobactam: cefepime and AAI101 as a potent β-Lactam–β-lactamase inhibitor combination. Antimicrob. Agents Chemother.63, e00105–e00119 (2019). CASPubMedPubMed CentralGoogle Scholar
- Gaibani, P. et al. In vivo evolution of resistant subpopulations of KPC-producing Klebsiella pneumoniae during ceftazidime/avibactam treatment. J. Antimicrob. Chemother.73, 1525–1529 (2018). CASPubMedGoogle Scholar
- Bush, K. Past and present perspectives on β-lactamases. Antimicrob. Agents Chemother.62, e01076-18 (2018). This study presents an extensive overview of β-lactamases including evolution and present situation. PubMedPubMed CentralGoogle Scholar
- Veeraraghavan, B. et al. Newer β-lactam/β-lactamase inhibitor for multidrug-resistant gram-negative infections: challenges, implications and surveillance strategy for India. Indian J. Med. Microbiol.36, 334–343 (2018). PubMedGoogle Scholar
- Negash, K. H., Norris, J. K. S. & Hodgkinson, J. T. Siderophore–antibiotic conjugate design: new drugs for bad bugs? Molecules24, 3314 (2019). CASPubMed CentralGoogle Scholar
- Ito, A. et al. Siderophore cephalosporin cefiderocol utilizes ferric iron transporter systems for antibacterial activity against Pseudomonas aeruginosa. Antimicrob. Agents Chemother.60, 7396–7401 (2016). CASPubMedPubMed CentralGoogle Scholar
- Jacobs, M. R. et al. ARGONAUT-I: Activity of cefiderocol (S-649266), a siderophore cephalosporin, against gram-negative bacteria, including carbapenem-resistant nonfermenters and Enterobacteriaceae with defined extended-spectrum β-lactamases and carbapenemases. Antimicrob. Agents Chemother.63, e01801–e01818 (2018). PubMedPubMed CentralGoogle Scholar
- Karlowsky, J. A. et al. In vitro activity of cefiderocol, a siderophore cephalosporin, against gram-negative bacilli isolated by clinical laboratories in North America and Europe in 2015–2016: SIDERO-WT-2015. Int. J. Antimicrob. Agents53, 456–466 (2019). CASPubMedGoogle Scholar
- Kazmierczak, K. M. et al. In vitro activity of cefiderocol, a siderophore cephalosporin, against a recent collection of clinically relevant carbapenem-non-susceptible Gram-negative bacilli, including serine carbapenemase- and metallo-β-lactamase-producing isolates (SIDERO-WT-2014 study). Int. J. Antimicrob. Agents53, 177–184 (2019). CASPubMedGoogle Scholar
- Echols, R., Ariyasu, M. & Nagata, T. D. Pathogen-focused clinical development to address unmet medical need: cefiderocol targeting carbapenem resistance. Clin. Infect. Dis.69, S559–S564 (2019). PubMedPubMed CentralGoogle Scholar
- Food and Drug Administration. FDA briefing document: meeting of the Antimicrobial Drugs Advisory Committee (AMDAC) https://www.fda.gov/media/131703/download (2019).
- Blais, J. et al. In vitro activity of LYS228, a novel monobactam antibiotic, against multidrug-resistant Enterobacteriaceae. Antimicrob. Agents Chemother.62, e00552–18 (2018). CASPubMedPubMed CentralGoogle Scholar
- Dean, C. R. et al. Mode of action of the monobactam LYS228 and mechanisms decreasing in vitro susceptibility in escherichia coli and Klebsiella pneumoniae. Antimicrob. Agents Chemother.62, e01200–e01218 (2018). CASPubMedPubMed CentralGoogle Scholar
- Dunne, M. Huband, M., Flamm, R., Aronin, S. & Puttagunta, S. Prediction of sulopenem activity against Enterobacteriaceae using ertapenem as a surrogate. ASM Microbe 2018https://d1io3yog0oux5.cloudfront.net/_1af31c1063e9451129263f05d42bc5fa/iterumtx/db/395/2664/pdf/ASM+2018_Ertapenem+surrogate.Final.pdf (2018).
- Iterum Therapeutics. Press release 10. 12. 2019: Iterum Therapeutics announces topline results from phase III clinical trial of oral and IV sulopenem for the treatment of complicated intra-abdominal infections. https://www.globenewswire.com/news-release/2019/12/10/1958907/0/en/Iterum-Therapeutics-Announces-Topline-Results-from-Phase-3-Clinical-Trial-of-Oral-and-IV-Sulopenem-for-the-Treatment-of-Complicated-Intra-abdominal-Infections.html (2019).
- Karlowsky, J. A. et al. In vitro activity of sulopenem, an oral penem, against urinary isolates of escherichia coli. Antimicrob. Agents Chemother.63, e01832-18 (2018). PubMedPubMed CentralGoogle Scholar
- McEntee, L. et al. Pharmacodynamics of tebipenem: new options for oral treatment of multidrug-resistant Gram-negative infections. Antimicrob. Agents Chemother.63, e00603–e00619 (2019). CASPubMedPubMed CentralGoogle Scholar
- Richter, S. E. et al. Risk factors for development of carbapenem resistance among gram-negative rods. Open Forum Infect. Dis.6, ofz027 (2019). PubMedPubMed CentralGoogle Scholar
- Thaker, M., Spanogiannopoulos, P. & Wright, G. D. The tetracycline resistome. Cell Mol. Life Sci.67, 419–431 (2010). This study presents an overview of structure, mechanism and regulation of the genes and proteins associated with tetracycline resistance. CASPubMedGoogle Scholar
- Villa, L., Feudi, C., Fortini, D., García-Fernández, A. & Carattoli, A. Genomics of KPC-producing Klebsiella pneumoniae sequence type 512 clone highlights the role of RamR and ribosomal S10 protein mutations in conferring tigecycline resistance. Antimicrob. Agents Chemother.58, 1707–1712 (2014). PubMedPubMed CentralGoogle Scholar
- Park, J. et al. Plasticity, dynamics, and inhibition of emerging tetracycline resistance enzymes. Nat. Chem. Biol.13, 730–736 (2017). CASPubMedPubMed CentralGoogle Scholar
- Sun, J. et al. Plasmid-encoded tet(X) genes that confer high-level tigecycline resistance in Escherichia coli. Nat. Microbiol.4, 1457–1464 (2019). CASPubMedPubMed CentralGoogle Scholar
- Grossman, T. H. Tetracycline antibiotics and resistance. Cold Spring Harb. Perspect. Med.6, a025387–a025387 (2016). PubMedPubMed CentralGoogle Scholar
- He, T. et al. Emergence of plasmid-mediated high-level tigecycline resistance genes in animals and humans. Nat. Microbiol.4, 1450–1456 (2019). CASPubMedGoogle Scholar
- Zhang, Y., Lin, X. & Bush, K. In vitro susceptibility of β-lactamase-producing carbapenem-resistant Enterobacteriaceae (CRE) to eravacycline. J. Antibiotics69, 600–604 (2016). CASGoogle Scholar
- Pfaller, M. A., Rhomberg, P. R., Huband, M. D. & Flamm, R. K. Activity of omadacycline tested against Enterobacteriaceae causing urinary tract infections from a global surveillance program (2014). Diagn. Microbiol. Infect. Dis.91, 179–183 (2018). CASPubMedGoogle Scholar
- Kaminishi, T. et al. Third-generation tetracycline KBP-7072 exploit reveal a new potential primary tetracycline binding pocket. Preprint at https://doi.org/10.1101/508218 (2018).
- Yang, F., Wang, Y., Wang, P., Hong, M. & Benn, V. Multiple ascending dose safety, tolerability, and pharmacokinetics of KBP-7072, a novel third-generation tetracycline. Open For. Infect. Dis.4, S291–S291 (2017). Google Scholar
- Huband, M. D. et al. Activity of KBP-7072 against recent and molecularly characterized Acinetobacter baumannii isolates. ASM/ESCMID Drug Development Conference 2019https://www.jmilabs.com/data/posters/ASM-ESCMID2019-KBP-7072-Acinetobacter.pdf (JMI Labs, 2019).
- Falagas, M. E. et al. Activity of TP-6076 against carbapenem-resistant Acinetobacter baumannii isolates collected from inpatients in Greek hospitals. Int. J. Antimicrob. Agents52, 269–271 (2018). CASPubMedGoogle Scholar
- Fyfe, C., LeBlanc, G., Close, B. & Newman J. TP-6076 is active against carbapenem- and polymyxin-resistant Enterobacteriaceae and Acinetobacter baumannii isolates. ECCMID 2017https://www.escmid.org/typo3conf/ext/escmid_solr/Resources/Images/icn_abstract-hover.png (Vienna, 2017).
- Sun, C. et al. TP-6076, a fully synthetic tetracycline antibacterial agent, is highly potent against a broad range of pathogens, including carbapenem-resistant Enterobacteriaceae. ASM Microbe 2017https://www.tphase.com/wp-content/uploads/2019/04/Sun-S_ASM_2017_SUN-332-poster_6076-SAR_FINAL.pdf (2017).
- Tsai, L. & Moore, A. Safety, tolerability, and pharmacokinetics of multiple doses of TP-6076, a novel, fully synthetic tetracycline, in a phase I study. Open Forum Infect. Dis.5, S420–S420 (2018). PubMed CentralGoogle Scholar
- Grossman, T. H. et al. Fluorocycline TP-271 is potent against complicated community-acquired bacterial pneumonia pathogens. mSphere2, e00004–e00017 (2017). CASPubMedPubMed CentralGoogle Scholar
- Aruhomukama, D., Sserwadda, I. & Mboowa, G. Investigating colistin drug resistance: the role of high-throughput sequencing and bioinformatics. F1000Research8, 150–150 (2019). PubMedPubMed CentralGoogle Scholar
- Brown, P. et al. Design of next generation polymyxins with lower toxicity: the discovery of SPR206. ACS Infect. Dis.5, 1645–1656 (2019). CASPubMedGoogle Scholar
- Corbett, D. et al. Potentiation of antibiotic activity by a novel cationic peptide: potency and spectrum of activity of SPR741. Antimicrob. Agents Chemother.61, e00200–e00217 (2017). CASPubMedPubMed CentralGoogle Scholar
- Fleischmann, W. A., Greenwood-Quaintance, K. E. & Patel, R. In vitro activity of plazomicin compared to amikacin, gentamicin, and tobramycin against multidrug resistant aerobic Gram-negative bacilli. Antimicrob. Agents Chemother. https://doi.org/10.1128/AAC.01711-19 (2019).
- Serio, A., Keepers, T., Andrews, L. & Krause, K. Aminoglycoside revival: review of a historically important class of antimicrobials undergoing rejuvenation. EcoSal Plushttps://doi.org/10.1128/ecosalplus.ESP-0002-2018 (2018).
- Livermore, D. M. et al. Activity of aminoglycosides, including ACHN-490, against carbapenem-resistant Enterobacteriaceae isolates. J. Antimicrob. Chemother.66, 48–53 (2010). PubMedGoogle Scholar
- Juhas, M. et al. In vitro activity of apramycin against multidrug-, carbapenem- and aminoglycoside-resistant Enterobacteriaceae and Acinetobacter baumannii. J. Antimicrob. Chemother.74, 944–952 (2019). CASPubMedPubMed CentralGoogle Scholar
- Hunter, J. E., Hart, C. A., Shelley, J. C., Walton, J. R. & Bennett, M. Human isolates of apramycin-resistant Escherichia coli which contain the genes for the AAC(3)IV enzyme. Epidemiol. Infect.110, 253–259 (1993). CASPubMedPubMed CentralGoogle Scholar
- Yang, Y., Hu, F. & Zhu, D. evaluation of contezolid activity to anaerobic and gram-positive-cocci isolates from a phase III acute bacterial skin and skin structure infection clinical trial (MRX-I-06). Open Forum Infect. Dis.6, S315–S315 (2019). PubMed CentralGoogle Scholar
- Flamm, R. K., Rhomberg, P. R. & Sader, H. S. In vitro activity of the novel lactone ketolide nafithromycin (WCK 4873) against contemporary clinical bacteria from a global surveillance program. Antimicrob. Agents Chemother.61, e01230–17 (2017). CASPubMedPubMed CentralGoogle Scholar
- Fisher, C. et al. Activity of TNP-2092 against biofilms formed by prosthetic joint infection-associated staphylococci. Open Forum Infect. Dis.6, S313–S313 (2019). PubMed CentralGoogle Scholar
- Yao, J., Maxwell, J. B. & Rock, C. O. Resistance to AFN-1252 arises from missense mutations in Staphylococcus aureus enoyl-acyl carrier protein reductase (FabI). J. Biol. Chem.288, 36261–36271 (2013). CASPubMedPubMed CentralGoogle Scholar
- Schiebel, J. et al. Staphylococcus aureus FabI: inhibition, substrate recognition, and potential implications for in vivo essentiality. Structure20, 802–813 (2012). CASPubMedPubMed CentralGoogle Scholar
- Hawser, S. et al. Activity of debio 1452 against Staphylococcus spp. collected in 2013/2014. ECCMID 2016https://www.ihma.com/app/uploads/P139_POSTER_Debiopharm_IHMA_ECCMID_2016_v1-final.pdf (2016).
- Menetrey, A. et al. Bone and joint tissue penetration of the Staphylococcus-selective antibiotic afabicin in patients undergoing elective hip replacement surgery. Antimicrob. Agents Chemother.63, e01669-18 (2019). PubMedPubMed CentralGoogle Scholar
- Escaich, S. et al. The MUT056399 Inhibitor of FabI is a new antistaphylococcal compound. Antimicrob. Agents Chemother.55, 4692–4697 (2011). CASPubMedPubMed CentralGoogle Scholar
- Kaul, M. et al. TXA709, an FtsZ-targeting benzamide prodrug with improved pharmacokinetics and enhanced in vivo efficacy against methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother.59, 4845 (2015). CASPubMedPubMed CentralGoogle Scholar
- Kaul, M., Mark, L., Parhi, A. K., LaVoie, E. J. & Pilch, D. S. Combining the FtsZ-targeting prodrug TXA709 and the cephalosporin cefdinir confers synergy and reduces the frequency of resistance in methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother.60, 4290 (2016). CASPubMedPubMed CentralGoogle Scholar
- Basarab, G. S. et al. Responding to the challenge of untreatable gonorrhea: ETX0914, a first-in-class agent with a distinct mechanism-of-action against bacterial type II topoisomerases. Sci. Rep.5, 11827–11827 (2015). CASPubMedPubMed CentralGoogle Scholar
- Taylor, S. N. et al. Gepotidacin for the treatment of uncomplicated urogenital gonorrhea: a phase II, randomized, dose-ranging, single-oral dose evaluation. Clin. Infect. Dis.67, 504–512 (2018). CASPubMedPubMed CentralGoogle Scholar
- Negash, K. et al. The metabolism and disposition of GSK2140944 in healthy human subjects. Xenobiotica46, 683–702 (2016). CASPubMedGoogle Scholar
- Flamm, R. K., Farrell, D. J., Rhomberg, P. R., Scangarella-Oman, N. E. & Sader, H. S. Gepotidacin (GSK2140944) in vitro activity against Gram-positive and Gram-negative bacteria. Antimicrob. Agents Chemother.61, e00468–17 (2017). CASPubMedPubMed CentralGoogle Scholar
- Bisacchi, G. S. & Manchester, J. I. A new-class antibacterial — almost. Lessons in drug discovery and development: a critical analysis of more than 50 years of effort toward ATPase inhibitors DNA gyrase topoisomerase IV. ACS Infect. Dis.1, 4–41 (2015). This study presents insights into the historical attempts to discover and develop ATPase inhibitors of gyrase and topo IV as new-class antibacterial agents with broad-spectrum potential. CASPubMedGoogle Scholar
- Brown-Elliott, B. A., Rubio, A. & Wallace, R. J. Jr. In vitro susceptibility testing of a novel benzimidazole, SPR719, against nontuberculous mycobacteria. Antimicrob. Agents Chemother.62, e01503–e01518 (2018). PubMedPubMed CentralGoogle Scholar
- Food & Drug Administration. Drug approval package: XENLENTA https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/211672Orig1s000,211673Orig1s000TOC.cfm (2020).
- Cho, J. C., Crotty, M. P. & Pardo, J. Ridinilazole: a novel antimicrobial for Clostridium difficile infection. Ann. Gastroenterol.32, 134–140 (2019). PubMedGoogle Scholar
- Bassères, E. et al. Understanding the Mechanism of Action of Ridinilazole (SMT19969), a Novel Treatment for Clostridium difficile. ECCMID 2016https://www.escmid.org/escmid_publications/escmid_elibrary/material/?mid=50079 (2016).
- Khalaf, A. I. et al. Distamycin analogues with enhanced lipophilicity: synthesis and antimicrobial activity. J. Med. Chem.47, 2133–2156 (2004). CASPubMedGoogle Scholar
- Garey, K. W. et al. A Randomized, blinded, placebo- and vancomycin-controlled, first-in-human (FIH) study of the safety, pharmacokinetics (PK), and fecal microbiome effects of ACX-362E, a novel anti-clostridial DNA polymerase IIIC (polIIIC) inhibitor. Open Forum Infect. Dis.6, S995–S996 (2019). PubMed CentralGoogle Scholar
- Green, L. S. et al. Inhibition of methionyl-tRNA synthetase by REP8839 and effects of resistance mutations on enzyme activity. Antimicrob. Agents Chemother.53, 86–94 (2009). CASPubMedGoogle Scholar
- Roberts, J. A. et al. Individualised antibiotic dosing for patients who are critically ill: challenges and potential solutions. Lancet Infect. Dis.14, 498–509 (2014). PubMedPubMed CentralGoogle Scholar
- Livermore, D. M. The 2018 Garrod Lecture: preparing for the black swans of resistance. J. Antimicrob. Chemother.73, 2907–2915 (2018). CASPubMedGoogle Scholar
- Amaral, L., Martins, A., Spengler, G. & Molnar, J. Efflux pumps of Gram-negative bacteria: what they do, how they do it, with what and how to deal with them. Front. Pharmacol.4, 168 (2014). PubMedPubMed CentralGoogle Scholar
Acknowledgements
The authors thank S. Paulin and P. Beyer (WHO) and the members of the advisory group of the WHO pipeline report — M. Butler, L. Czaplewski, J. Hood, F. Franceschi, R. Kozlov, C. Lienhardt, N. Ohmagari, L. Silver and R. Alm — for their support, advice and contributions.
Author information
Authors and Affiliations
- Center for Anti-Infective Agents, Vienna, Austria Ursula Theuretzbacher
- Department of Biology, Indiana University, Bloomington, IN, USA Karen Bush
- Infection Control Program, World Health Organization Collaborating Centre on Patient Safety, Geneva University Hospitals and Faculty of Medicine, Geneva, Switzerland Stephan Harbarth
- Diseases Institute, Rambam Health Care Campus. Ruth and Bruce Rappaport Faculty of Medicine, Technion – Israel Institute of Technology, Haifa, Israel Mical Paul
- F2G Ltd., Wellesley Hills, MA,, USA John H. Rex
- Infectious Diseases, Department of Diagnostic and Public Health, Verona University Hospital, Verona, Italy Evelina Tacconelli
- Oxford University Clinical Research Unit, Ho Chi Minh City, Vietnam Guy E. Thwaites
- Centre for Tropical Medicine and Global Health, Nuffield Department of Medicine, University of Oxford, Oxford, UK Guy E. Thwaites
- Ursula Theuretzbacher
